Show code cell source
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
from sklearn.preprocessing import StandardScaler, MinMaxScaler
from sklearn.model_selection import train_test_split
from sklearn.linear_model import LinearRegression
from rdkit import Chem
from rdkit.Chem import (
PandasTools,
Descriptors
)
from rdkit.Chem.Draw import IPythonConsole
Notebook - ML for thermochemical data prediction#
We can use the data you worked with for the EDA workshop to demonstrate how fit a simple ML model to predict the boiling point of organic compounds from a small set of descriptors.
This is an example of
Supervised learning as applied to a regression task.
- Supervised learning
The model learns from data for which a label (known value or category) is associated with each input (set of feature values)
- Regression task
Model predicts a continuous numerical value based on the input features
bp_df = pd.read_csv("data/alcohol_acid_phys_data_cleaned.csv")
bp_df
| Class | IUPAC name | #C | #H | #O | Molweight g/mol | mp / dC | bp / dC | density / g/ml | SMILES | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1-alkanol | Methanol | 1 | 4 | 1 | 32.04 | -98.0 | 65 | 0.7910 | CO |
| 1 | 1-alkanol | Ethanol | 2 | 6 | 1 | 46.07 | -114.0 | 78 | 0.7890 | CCO |
| 2 | 1-alkanol | 1-Propanol | 3 | 8 | 1 | 60.09 | -124.0 | 97 | 0.8040 | CCCO |
| 3 | 1-alkanol | 1-Butanol | 4 | 10 | 1 | 74.12 | -89.0 | 118 | 0.8100 | CCCCO |
| 4 | 1-alkanol | 1-Pentanol | 5 | 12 | 1 | 88.15 | -78.0 | 138 | 0.8140 | CCCCCO |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 73 | Branched alkanoic acid | 4-Methylpentanoic acid | 6 | 12 | 2 | 116.16 | -33.0 | 200 | 0.9230 | CC(C)CCC(O)=O |
| 74 | Benzoic acids | Benzoic acid | 7 | 6 | 2 | 122.12 | 122.0 | 249 | 1.2660 | OC(=O)c1ccccc1 |
| 75 | Benzoic acids | 2-Methyl-benzoic acid | 8 | 8 | 2 | 136.14 | 107.0 | 258 | 1.0620 | Cc1ccccc1C(O)=O |
| 76 | Phenyl- alkanoic acid | Phenylethanoic acid | 8 | 8 | 2 | 136.14 | 77.0 | 266 | 1.2286 | OC(=O)Cc1ccccc1 |
| 77 | Alkanedioic acid | Decanedioic acid | 10 | 18 | 4 | 202.24 | 131.0 | 374 | 1.2710 | OC(=O)CCCCCCCCC(O)=O |
78 rows × 10 columns
The data is read in from a csv file of the data cleaned in the EDA workshop, so should be prepared, but we can quickly check.
# Check for missing values
bp_df.isna().sum()
Class 0
IUPAC name 0
#C 0
#H 0
#O 0
Molweight g/mol 0
mp / dC 0
bp / dC 0
density / g/ml 0
SMILES 0
dtype: int64
# Check data types
bp_df.info()
<class 'pandas.core.frame.DataFrame'>
RangeIndex: 78 entries, 0 to 77
Data columns (total 10 columns):
# Column Non-Null Count Dtype
--- ------ -------------- -----
0 Class 78 non-null object
1 IUPAC name 78 non-null object
2 #C 78 non-null int64
3 #H 78 non-null int64
4 #O 78 non-null int64
5 Molweight g/mol 78 non-null float64
6 mp / dC 78 non-null float64
7 bp / dC 78 non-null int64
8 density / g/ml 78 non-null float64
9 SMILES 78 non-null object
dtypes: float64(3), int64(4), object(3)
memory usage: 6.2+ KB
The numerical types are as expected. The category type for the class column has not been automatically recognised, understandably. For now we will not be working with that column, so will leave as is.
We will start by dropping any rows without a SMILES string and also the melting point column as we are going to try to predict the boiling point.
# Drop rows with missing SMILES string
bp_df = bp_df[bp_df["SMILES"] != "not found"].reset_index(drop=True)
bp_df = bp_df.drop(columns=["mp / dC"])
bp_df
| Class | IUPAC name | #C | #H | #O | Molweight g/mol | bp / dC | density / g/ml | SMILES | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | 1-alkanol | Methanol | 1 | 4 | 1 | 32.04 | 65 | 0.7910 | CO |
| 1 | 1-alkanol | Ethanol | 2 | 6 | 1 | 46.07 | 78 | 0.7890 | CCO |
| 2 | 1-alkanol | 1-Propanol | 3 | 8 | 1 | 60.09 | 97 | 0.8040 | CCCO |
| 3 | 1-alkanol | 1-Butanol | 4 | 10 | 1 | 74.12 | 118 | 0.8100 | CCCCO |
| 4 | 1-alkanol | 1-Pentanol | 5 | 12 | 1 | 88.15 | 138 | 0.8140 | CCCCCO |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 72 | Branched alkanoic acid | 4-Methylpentanoic acid | 6 | 12 | 2 | 116.16 | 200 | 0.9230 | CC(C)CCC(O)=O |
| 73 | Benzoic acids | Benzoic acid | 7 | 6 | 2 | 122.12 | 249 | 1.2660 | OC(=O)c1ccccc1 |
| 74 | Benzoic acids | 2-Methyl-benzoic acid | 8 | 8 | 2 | 136.14 | 258 | 1.0620 | Cc1ccccc1C(O)=O |
| 75 | Phenyl- alkanoic acid | Phenylethanoic acid | 8 | 8 | 2 | 136.14 | 266 | 1.2286 | OC(=O)Cc1ccccc1 |
| 76 | Alkanedioic acid | Decanedioic acid | 10 | 18 | 4 | 202.24 | 374 | 1.2710 | OC(=O)CCCCCCCCC(O)=O |
77 rows × 9 columns
# Add RDKit molecules to the dataframe
PandasTools.AddMoleculeColumnToFrame(bp_df, smilesCol="SMILES")
bp_df
| Class | IUPAC name | #C | #H | #O | Molweight g/mol | bp / dC | density / g/ml | SMILES | ROMol | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1-alkanol | Methanol | 1 | 4 | 1 | 32.04 | 65 | 0.7910 | CO |  |
| 1 | 1-alkanol | Ethanol | 2 | 6 | 1 | 46.07 | 78 | 0.7890 | CCO |  |
| 2 | 1-alkanol | 1-Propanol | 3 | 8 | 1 | 60.09 | 97 | 0.8040 | CCCO |  |
| 3 | 1-alkanol | 1-Butanol | 4 | 10 | 1 | 74.12 | 118 | 0.8100 | CCCCO |  |
| 4 | 1-alkanol | 1-Pentanol | 5 | 12 | 1 | 88.15 | 138 | 0.8140 | CCCCCO |  |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 72 | Branched alkanoic acid | 4-Methylpentanoic acid | 6 | 12 | 2 | 116.16 | 200 | 0.9230 | CC(C)CCC(O)=O |  |
| 73 | Benzoic acids | Benzoic acid | 7 | 6 | 2 | 122.12 | 249 | 1.2660 | OC(=O)c1ccccc1 |  |
| 74 | Benzoic acids | 2-Methyl-benzoic acid | 8 | 8 | 2 | 136.14 | 258 | 1.0620 | Cc1ccccc1C(O)=O |  |
| 75 | Phenyl- alkanoic acid | Phenylethanoic acid | 8 | 8 | 2 | 136.14 | 266 | 1.2286 | OC(=O)Cc1ccccc1 |  |
| 76 | Alkanedioic acid | Decanedioic acid | 10 | 18 | 4 | 202.24 | 374 | 1.2710 | OC(=O)CCCCCCCCC(O)=O |  |
77 rows × 10 columns
Show code cell source
# Adapted from https://greglandrum.github.io/rdkit-blog/posts/2022-12-23-descriptor-tutorial.html
def getMolDescriptors(mol, descriptor_list=None, missingVal=None):
''' calculate the full list of descriptors for a molecule
missingVal is used if the descriptor cannot be calculated
'''
res = {}
if not(descriptor_list):
descriptors = Descriptors._descList
# TODO: Add else clause to handle a list numbers corresponding to the descriptor indices
else:
descriptors = [Descriptors._descList[idx] for idx in descriptor_list]
for nm,fn in descriptors:
# some of the descriptor fucntions can throw errors if they fail, catch those here:
try:
val = fn(mol)
except:
# print the error message:
import traceback
traceback.print_exc()
# and set the descriptor value to whatever missingVal is
val = missingVal
res[nm] = val
return res
# These are the descriptors selected to calculate for the molecules.
# 118 NumHAcceptors
# 119 NumHDonors
# 27 BalabanJ - a topological descriptor expressing molecular connectivity and branching
# 28 BertzCT - a topological complexity index
# 83 TPSA - total polar surface area
descriptor_list = [118, 119, 27, 28, 83]
calc_descriptors = [getMolDescriptors(mol, descriptor_list=descriptor_list) for mol in bp_df["ROMol"]]
# Create a dataframe from the calculated descriptors
descriptor_df = pd.DataFrame(calc_descriptors)
# Add the descriptors to the dataframe as new columns
bp_df = pd.concat([bp_df, descriptor_df], axis=1)
bp_df
| Class | IUPAC name | #C | #H | #O | Molweight g/mol | bp / dC | density / g/ml | SMILES | ROMol | NumHAcceptors | NumHDonors | BalabanJ | BertzCT | TPSA | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1-alkanol | Methanol | 1 | 4 | 1 | 32.04 | 65 | 0.7910 | CO | <rdkit.Chem.rdchem.Mol object at 0x154fd8c10> | 1 | 1 | 1.000000 | 2.000000 | 20.23 |
| 1 | 1-alkanol | Ethanol | 2 | 6 | 1 | 46.07 | 78 | 0.7890 | CCO | <rdkit.Chem.rdchem.Mol object at 0x154fd8eb0> | 1 | 1 | 1.632993 | 2.754888 | 20.23 |
| 2 | 1-alkanol | 1-Propanol | 3 | 8 | 1 | 60.09 | 97 | 0.8040 | CCCO | <rdkit.Chem.rdchem.Mol object at 0x154fd8f90> | 1 | 1 | 1.974745 | 5.245112 | 20.23 |
| 3 | 1-alkanol | 1-Butanol | 4 | 10 | 1 | 74.12 | 118 | 0.8100 | CCCCO | <rdkit.Chem.rdchem.Mol object at 0x154fd8f20> | 1 | 1 | 2.190610 | 11.119415 | 20.23 |
| 4 | 1-alkanol | 1-Pentanol | 5 | 12 | 1 | 88.15 | 138 | 0.8140 | CCCCCO | <rdkit.Chem.rdchem.Mol object at 0x154fd9000> | 1 | 1 | 2.339092 | 15.900135 | 20.23 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 72 | Branched alkanoic acid | 4-Methylpentanoic acid | 6 | 12 | 2 | 116.16 | 200 | 0.9230 | CC(C)CCC(O)=O | <rdkit.Chem.rdchem.Mol object at 0x154fdaea0> | 1 | 1 | 3.050078 | 76.597721 | 37.30 |
| 73 | Benzoic acids | Benzoic acid | 7 | 6 | 2 | 122.12 | 249 | 1.2660 | OC(=O)c1ccccc1 | <rdkit.Chem.rdchem.Mol object at 0x154fdaf10> | 1 | 1 | 2.981455 | 203.415953 | 37.30 |
| 74 | Benzoic acids | 2-Methyl-benzoic acid | 8 | 8 | 2 | 136.14 | 258 | 1.0620 | Cc1ccccc1C(O)=O | <rdkit.Chem.rdchem.Mol object at 0x154fdaf80> | 1 | 1 | 3.152941 | 253.189433 | 37.30 |
| 75 | Phenyl- alkanoic acid | Phenylethanoic acid | 8 | 8 | 2 | 136.14 | 266 | 1.2286 | OC(=O)Cc1ccccc1 | <rdkit.Chem.rdchem.Mol object at 0x154fdaff0> | 1 | 1 | 2.674242 | 215.959017 | 37.30 |
| 76 | Alkanedioic acid | Decanedioic acid | 10 | 18 | 4 | 202.24 | 374 | 1.2710 | OC(=O)CCCCCCCCC(O)=O | <rdkit.Chem.rdchem.Mol object at 0x154fdb060> | 2 | 2 | 2.989544 | 156.960812 | 74.60 |
77 rows × 15 columns
# To remind us of the relationship between the variables, we can plot a pairplot and heatmap
# Pairplot
sns.pairplot(bp_df, hue="Class", diag_kind="kde", palette="viridis")
plt.show()

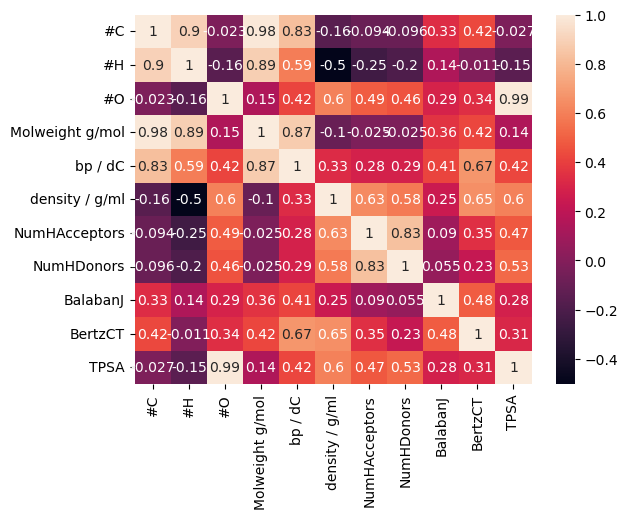
corr = bp_df.drop(columns=["Class", "IUPAC name", "SMILES", "ROMol"]).corr()
sns.heatmap(corr, annot=True)
plt.show()
# corr_abs = bp_df.drop(columns=["Class", "IUPAC name", "SMILES", "ROMol"]).corr().abs()
# upper = corr_abs.where(np.triu(np.ones(corr.shape), k=1).astype(bool))
# upper
This is all looking rather busy - it would be better to look at subsets of the features.
What we can see is:
The feature most strongly correlated with the target variable is the molecular weight.
Molecular weight is very strongly correlated with number of carbons and hydrogens
Most of the other features are only moderately or weakly correlated to the target variable.
# A closer look at the correlation between the boiling point and the molecular weight
sns.regplot(data=bp_df, x="Molweight g/mol", y="bp / dC", fit_reg=True, ci=None)
plt.show()
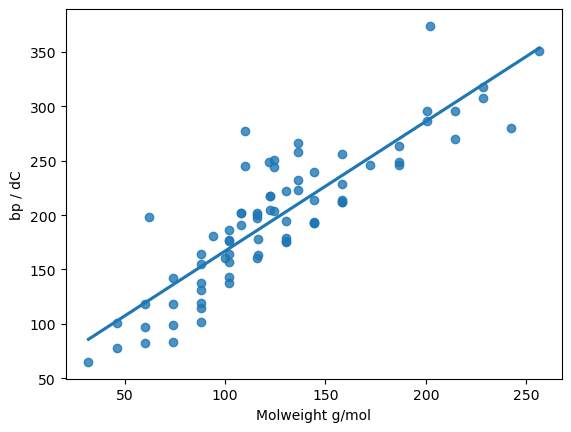
For the moment, we will drop the #C and #H columns. We will see that analysing the initial model can help tell us about the importance of the features.
We will also drop the non-numerical features for this task.
cols_drop = ["#C", "#H", "IUPAC name", "SMILES", "ROMol", "Class"]
prep_df = bp_df.drop(columns=cols_drop)
prep_df
| #O | Molweight g/mol | bp / dC | density / g/ml | NumHAcceptors | NumHDonors | BalabanJ | BertzCT | TPSA | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 32.04 | 65 | 0.7910 | 1 | 1 | 1.000000 | 2.000000 | 20.23 |
| 1 | 1 | 46.07 | 78 | 0.7890 | 1 | 1 | 1.632993 | 2.754888 | 20.23 |
| 2 | 1 | 60.09 | 97 | 0.8040 | 1 | 1 | 1.974745 | 5.245112 | 20.23 |
| 3 | 1 | 74.12 | 118 | 0.8100 | 1 | 1 | 2.190610 | 11.119415 | 20.23 |
| 4 | 1 | 88.15 | 138 | 0.8140 | 1 | 1 | 2.339092 | 15.900135 | 20.23 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 72 | 2 | 116.16 | 200 | 0.9230 | 1 | 1 | 3.050078 | 76.597721 | 37.30 |
| 73 | 2 | 122.12 | 249 | 1.2660 | 1 | 1 | 2.981455 | 203.415953 | 37.30 |
| 74 | 2 | 136.14 | 258 | 1.0620 | 1 | 1 | 3.152941 | 253.189433 | 37.30 |
| 75 | 2 | 136.14 | 266 | 1.2286 | 1 | 1 | 2.674242 | 215.959017 | 37.30 |
| 76 | 4 | 202.24 | 374 | 1.2710 | 2 | 2 | 2.989544 | 156.960812 | 74.60 |
77 rows × 9 columns
from sklearn.model_selection import train_test_split
from sklearn.preprocessing import MinMaxScaler
# By convention, the target variable is denoted by y and the features are denoted by X
y_full = prep_df["bp / dC"]
X_full = prep_df.drop(columns=["bp / dC"])
# train_test_split shuffles the data by default and splits it into training and testing sets. The
# proportion of the data that is used for testing is determined by the test_size parameter. Here,
# we are using 80 % of the data for training and 20% for the test set.
# The random_state parameter is used to set the seed for the random number generator so that the
# results are reproducible.
X_train, X_test, y_train, y_test = train_test_split(X_full, y_full, test_size=0.2, random_state=42)
# Check the size of the training and testing sets
X_train.shape, X_test.shape, y_train.shape, y_test.shape
((61, 8), (16, 8), (61,), (16,))
Scikit-learn makes many models available via a consistent interface.
We are going to use a linear regression model for this task.
from sklearn import linear_model
# Create a linear regression model
reg = linear_model.LinearRegression()
# Fit the model to the training data
reg.fit(X_train, y_train)
LinearRegression()In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook.
On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
LinearRegression()
# Predict the boiling points of the test set
y_pred = reg.predict(X_test)
We can plot the boiling points that the model predicted for the test set against the known true values to see how good a job the model makes of the predictions
plt.scatter(y_test, y_pred)
plt.plot([y_test.min(), y_test.max()], [y_test.min(), y_test.max()], '-', lw=1, color="red")
plt.xlabel("True Values [bp / dC]")
plt.ylabel("Predictions [bp / dC]")
plt.show()
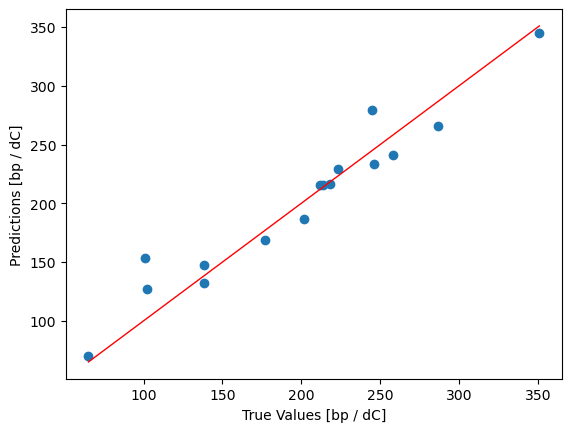
# The r^2 value is a measure of how well the model fits the data. It ranges from 0 to 1,
# with 1 indicating a perfect fit.
r2 = reg.score(X_test, y_test)
r2
0.9287935935680889
If we look back, we can see that the Pearson correlation coefficient for the relationship between molecular weight and boiling point was 0.84, so the model predicts more accurately than using the molecular weight alone.
The model coefficients tell us about the weighting of the features used by the fitted model.
print(X_full.columns)
reg.coef_
Index(['#O', 'Molweight g/mol', 'density / g/ml', 'NumHAcceptors',
'NumHDonors', 'BalabanJ', 'BertzCT', 'TPSA'],
dtype='object')
array([-1.76222475e+01, 1.17239618e+00, 1.64551206e+02, -2.42988355e+00,
2.57293551e+01, -6.34194132e+00, 5.78435772e-02, 1.26142804e+00])
However, because the magnitude of the features’ values are on different scales, the coefficients also incorporate the different scales.
A scaler can be used to transform the features to a consistent scale. Here’s we’ll use a MinMaxScaler to transform the features to have a scale between 0 and 1.
# Split the scaled data into training and testing sets
X_train, X_test, y_train, y_test = train_test_split(X_full, y_full, test_size=0.2, random_state=42)
# create a min/max scaler
scaler = MinMaxScaler()
# fit the scaler to the training data
scaler.fit(X_train)
# transform the training and testing data separately
scaled_X_train = scaler.transform(X_train)
scaled_X_test = scaler.transform(X_test)
# Create a linear regression model
reg = linear_model.LinearRegression()
# Fit the model to the training data
reg.fit(scaled_X_train, y_train)
# Predict the boiling points of the test set
y_pred = reg.predict(scaled_X_test)
plt.scatter(y_test, y_pred)
plt.plot([y_test.min(), y_test.max()], [y_test.min(), y_test.max()], '-', lw=1, color="red")
plt.xlabel("True Values [bp / dC]")
plt.ylabel("Predictions [bp / dC]")
plt.show()
# calculate the R^2 score
r2 = reg.score(scaled_X_test, y_test)
r2
0.9287935935680927
The model’s predictions look the same as before, but we can now look at the coefficients.
print(X_full.columns)
reg.coef_
Index(['#O', 'Molweight g/mol', 'density / g/ml', 'NumHAcceptors',
'NumHDonors', 'BalabanJ', 'BertzCT', 'TPSA'],
dtype='object')
array([ 1.65984399, 230.16481816, 81.1237445 , -10.77763669,
37.44176379, -13.68670374, 12.45958115, 10.69260041])
We can now see that the coefficients which represent the weights of the features in the fitted model indicate that molecular weight - as expected - and density are contributing most strongly to the model